Dear US customers please visit us on separate website Phenion-us.com
Switch to US site here
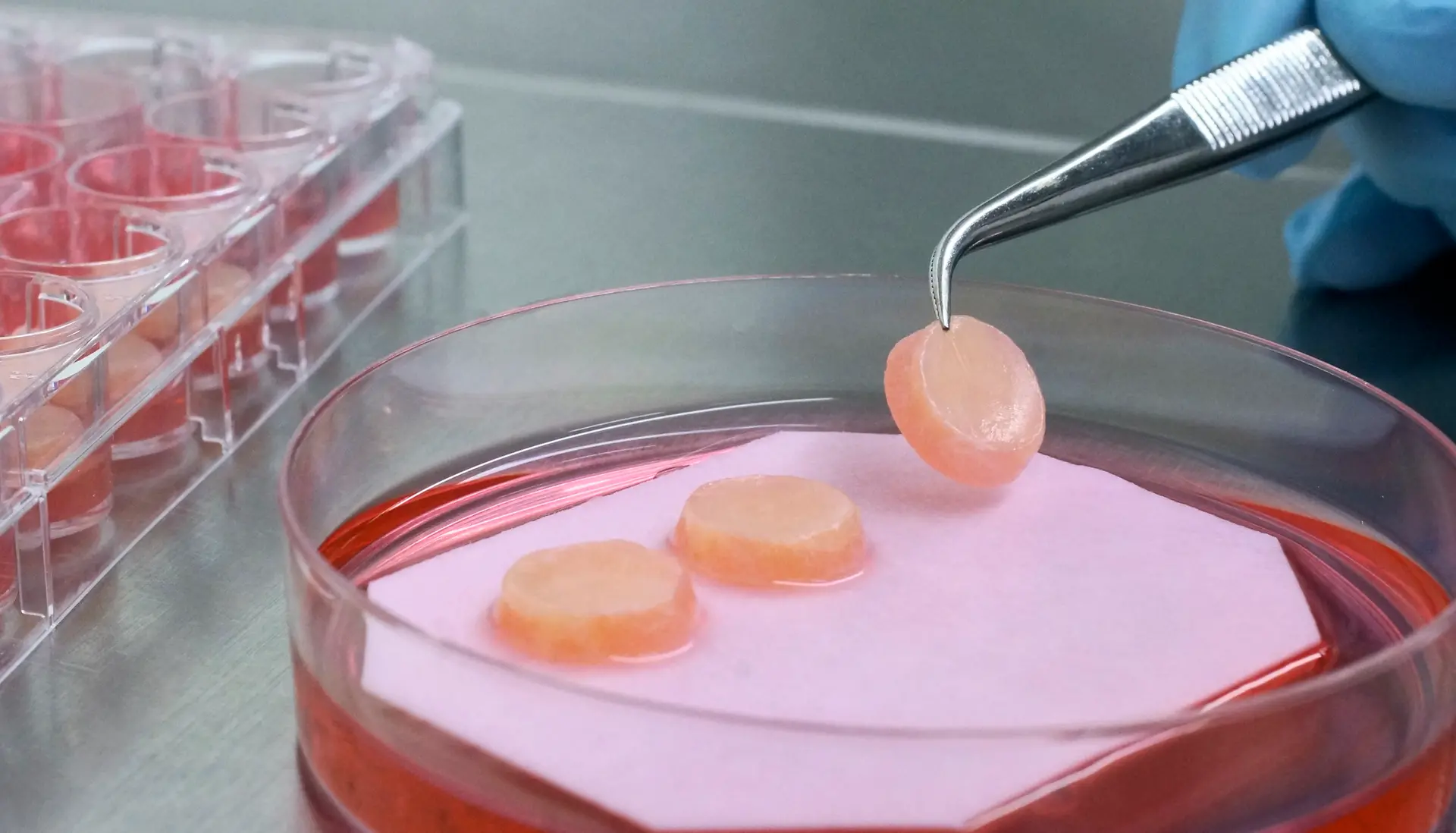
We provide worldwide best in class tissue engineered 3D skin models. Our artificial skin fulfills diverse in vitro research needs.
Phenion, a brand from Henkel, is a synonym for more than 15 years of state-of-the-art expertise in the development of alternatives to animal testing, in vitro research and tissue engineering. Find our products, e.g. artificial skin models build of both epidermis and dermis with a connective tissue made of collagen as well as of de novo synthesized extracellular matrix (ECM). Download our references and further information in our Information Center!
Building on a strong legacy of more than 145 years, we are leading the way to reimagine and improve life every day. Today and for generations to come. Through innovative and sustainable brands and technologies, across our teams around the world.
Henkel holds leading positions in both industrial and consumer businesses: Our portfolio includes well-known hair care products, laundry detergents, fabric softeners as well as adhesives, sealants, and functional coatings.






